溶血性贫血(HA)是因红细胞破坏加速、寿命缩短,超出骨髓造血代偿能力而引发的一类贫血性疾病。其临床表现多样,发病机制复杂,涉及红细胞内在缺陷与外在因素的相互作用。天津医科大学总医院付蓉教授在近期山东省青岛市召开的中国医疗保健国际交流促进会血液学分会2025年学术年会暨“华佗工程”MDT研讨会上的规范化诊疗专场中,带来了《溶血性贫血的诊断与鉴别》的专题发言,围绕溶血性贫血的分类、临床表现、病因机制、诊断策略及鉴别要点展开系统阐述,本刊特别整理课件内容,以飨读者。

分类与临床表现
溶血性贫血的核心特征是红细胞破坏加速,根据发病缓急可分为急性HA与慢性HA;急性HA是指由各种因素导致红细胞在短时间内的大量溶解,超过机体代偿能力所发生的一种贫血。临床表现为急性起病,全身不适,高热、寒战、头痛、腰酸背痛、腹痛,贫血,黄疸、尿呈酱油色或红葡萄酒色。慢性HA具有起病缓慢,症状轻微,有贫血、黄疸、肝脾肿大三大特征。慢性溶血性贫血患者由于长期的高胆红素血症可并发胆石症和肝功能损害等表现。
依据溶血场所分为血管内HA与血管外HA;血管内HA是指血细胞在血循环中溶破,血红蛋白直接释入血浆,又称细胞外溶血;正常衰老红细胞有10%~20%以此方法破坏。患者病因常为获得性膜缺陷,多为阵发性睡眠性血红蛋白尿(PNH),患者常具有酱油色或浓茶色尿。血管外HA是指由于红细胞膜表面的变化,被肝和脾的巨噬细胞辨认捕捉,在巨噬细胞内破坏,又称细胞内溶血;正常衰老红细胞80%~90%以此方法破坏。此类常见的病因为遗传性膜缺陷(遗传性球形细胞增多症)和遗传性血红蛋白病(珠蛋白生成障碍性贫血/地中海贫血)。
而按照红细胞破坏原因,可归为红细胞(RBC)内在缺陷HA与RBC外在因素HA两大类。内在缺陷源于红细胞膜、酶或血红蛋白的异常,多为遗传性,如遗传性球形红细胞增多症(HS)、葡萄糖-6-磷酸脱氢酶(G-6-PD)缺乏症等;外在因素则包括免疫性、机械性、感染或药物等获得性因素,如自身免疫性溶血性贫血(AIHA)、微血管病性溶血性贫血(MAHA)等。不同分类体系相互交织,共同构成了溶血性贫血的复杂疾病谱。
病因与发病机制
01
红细胞内在缺陷
01、膜缺陷性疾病
红细胞膜异常会导致红细胞稳定性下降,易于发生溶血。遗传性球形红细胞增多症(HS)是红细胞先天性膜缺陷引起的HA中最常见的⼀种类型,呈常染色体显性遗传,因锚定蛋白(如Ankyrin-1、Band-3)缺乏导致红细胞膜稳定性下降,易在脾脏被破坏。典型的临床表现有贫血、黄疸、RET增多、脾大,常伴胆石症。特征为血涂片见球形红细胞增多,可占红细胞的20%~40%,少数可达到80%以上。脾切除可治愈或缓解绝大多数患者的贫血,但无法逆转球形红细胞增多。通过EMA(伊红-5-马来酰亚胺)流式检测可辅助诊断,常见的基因突变包括ANK1、SLC4A1、SPTB等,不同基因突变对应的HS病例比例及严重程度各异。
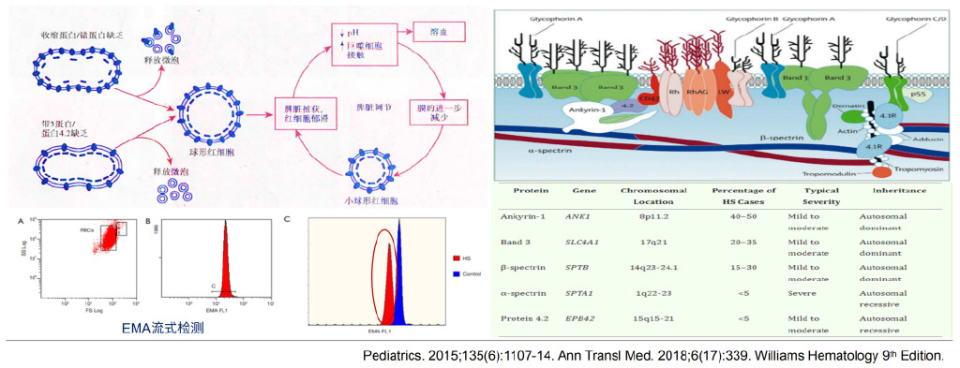
02、酶缺陷性疾病
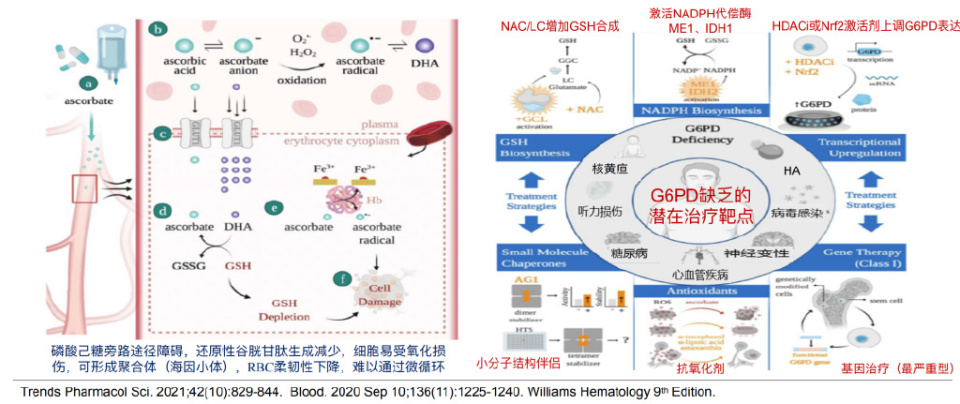
红细胞内多种酶缺陷会影响其代谢和功能,导致内环境失衡,引发溶血。G-6-PD缺乏症是最常见的红细胞酶缺陷病,由调控G-6-PD的基因突变所致,有多种基因变异型,呈X连锁不完全显性遗传。G-6-PD缺乏使红细胞易受氧化损伤,可导致急性溶血,具有自限性,需避免诱发溶血药物。其发病机制与磷酸己糖旁路途径障碍、还原性谷胱甘肽生成减少有关,细胞易受氧化损伤并形成海因小体,红细胞柔韧性下降。
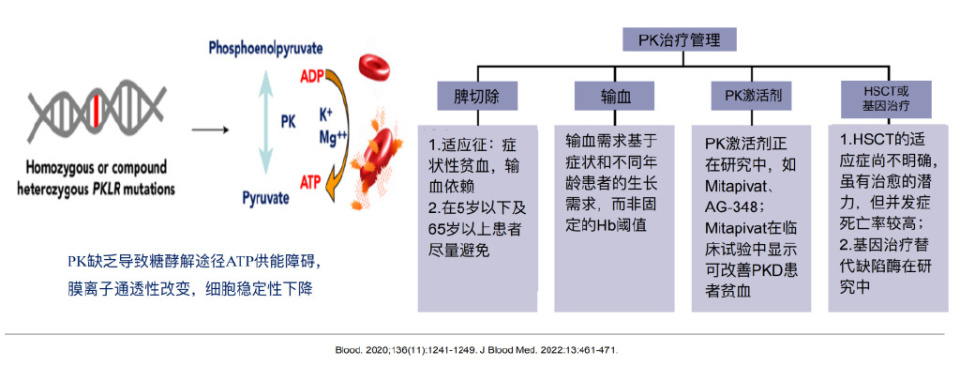
丙酮酸激酶缺乏症(PKD)是⼀种少见的常染色体隐性遗传病,因糖酵解途径ATP生成不足,导致膜离子通透性改变,细胞稳定性下降,表现为慢性溶血,具有异质性。常见并发症包括脾肿大、铁过载、胆石症等。诊断包括PK酶活性降低、PKLR基因突变(纯合或复合杂合型)、排除其他先天性HA等。治疗以支持治疗为主(如输血、祛铁),脾切除、PK激活剂(如Mitapivat)以及HSCT或基因治疗是潜在的治疗手段。
03、血红蛋白(HB)异常
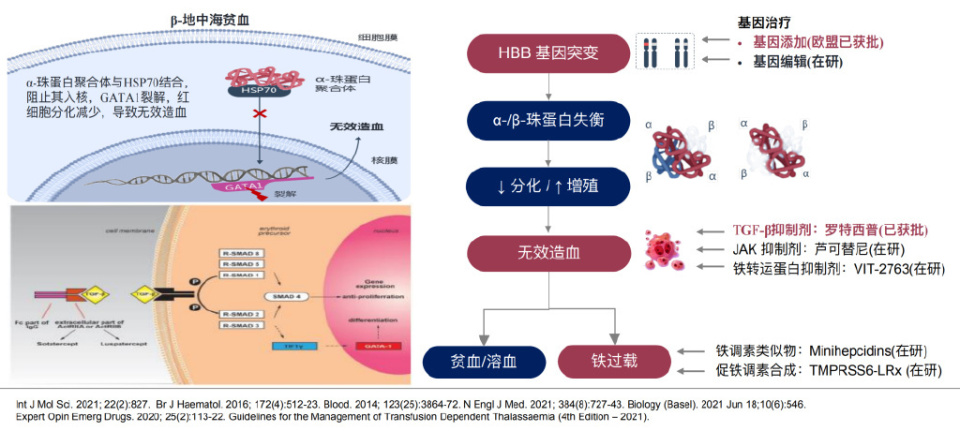
HB结构或合成异常会导致红细胞形态和功能改变,引发溶血。β-地中海贫血是一种遗传性小细胞性溶血性贫血,是由于β珠蛋白基因缺陷或突变,导致β珠蛋白链合成不足,而与之相匹配的α链过剩在红细胞内沉积,引起慢性溶血和脾大、无效造血和血红蛋白合成总体减少,产生小细胞低色素性红细胞和靶形红细胞以及血液中胎儿血红蛋白(HbF)和血红蛋白A2(HbA2)增高与血红蛋白A(HbA)减少或缺失的遗传性溶血性贫血。在β-地中海贫血中,α-珠蛋白聚合体聚集以及Smad2/3信号通路异常增强,均导致红细胞前体中GATA-1水平降低,加重无效造⾎。基因治疗(如基因添加、编辑技术)及靶向药物(如TGF-β抑制剂罗特西普、JAK抑制剂芦可替尼、铁转运蛋⽩抑制剂VIT-2763)为重型患者带来新希望。
04、锚定蛋白异常
PNH是一种由位于X染色体上的PIG-A基因突变引起的后天获得性造血干细胞克隆性疾病。其病理缺陷是糖基磷脂酰肌醇(GPI)合成异常,导致由GPI锚连在红细胞表面的一组锚连蛋白缺失,使红细胞在补体攻击下发生溶血,临床主要表现为溶血、骨髓衰竭和高风险并发血栓等。PNH患者的红细胞表面锚连蛋白(CD55、CD59等)缺失,导致红细胞在补体攻击下发生持续溶血,引起贫血和一氧化氮(NO)耗竭。
天津医科大学总医院作为国内PNH研究的重要临床中心,多年来对其发病机制进行了较为深入的研究,形成了系统的理论,包括二次基因突变、免疫逃逸、血栓并发症机制等,发表20余篇论著,其中SCI 15篇,获国家自然科学基金3项、天津市自然科学基金面上项目1项。在国内首次成功构建PNH小鼠模型,为探索新的靶向治疗提供动物模型,推动基础科研成果向临床应用的合理转化。
该中心多年来在PNH领域成果丰硕,贡献显著,包括:牵头中华医学会血液学分会“PNH卓越诊疗中心”建设项目,牵头中国阵发性睡眠性血红蛋白尿症登记项目,参与成立阵发性睡眠性血红蛋白尿症协作组,牵头发布中国首部《中国PNH患者生存状况白皮书》,牵头并主笔《中国PNH诊治指南》,率先在全国建立PNHHIS系统可视化筛查体系等。
在临床实践的探索上,该中心应用全外显子组测序(WES)技术识别PNH关键突变基因,为精准诊断提供技术支撑,推动诊断向分子层面深入;深入探究NK细胞在PNH免疫逃逸中的作用,揭示免疫机制奥秘,为免疫治疗开拓新思路,有望突破传统治疗局限。该中心还与南开大学生命科学院、天津医科大学基础医学院展开合作,基于基因芯片检测结果,探索经典型PNH新的分层治疗策略。
02
红细胞外在因素
01、免疫性因素
自身免疫性溶血性贫血(AIHA)因免疫系统紊乱产生自身抗体和/或补体破坏红细胞,所导致的获得性溶⾎性贫血。
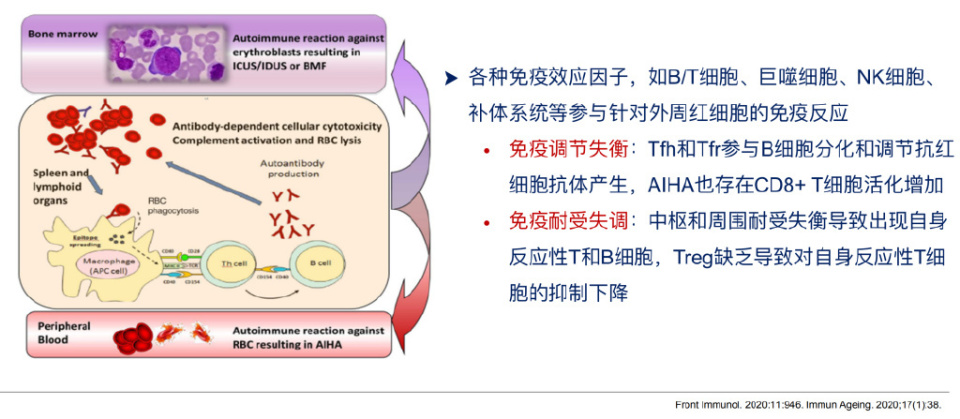
依据病因明确与否,分为继发性和原发性两类;依据自身抗体与红细胞结合所需的最适温度分为温抗体型、冷抗体型(包括冷凝集素综合征[CAS]及阵发性冷性血红蛋白尿症[PCH])和混合型;依据红细胞自身抗体检测结果,分为自身抗体阳性型和自身抗体阴性型。自身抗体阴性型AIHA临床症状符合溶血性贫血,除外其他溶血性贫血而免疫抑制治疗有效时可以诊断。其中,温抗体型AIHA(wAIHA)抗体类型主要为多克隆性IgG,最适反应温度为37℃。原发性wAIHA通常涉及以下几种溶血机制:红细胞膜上因吸附自身IgG抗体而被致敏,巨噬细胞表面的FcR与IgG的Fc片段结合,导致红细胞破坏或部分红细胞膜被吞噬,形成不易变形的球形红细胞,进入脾脏后被困于脾窦,进而被吞噬清除造成溶血;此外,IgG可以激活补体系统产生C3b,C3b调理的红细胞被肝脏的巨噬细胞捕获吞噬;在严重的情况下,补体激活会导致膜攻击体复合物(C5b-9)的形成并产生血管内溶血,但这种情况比较少见。
Immun Ageing. 2020;17(1):38.
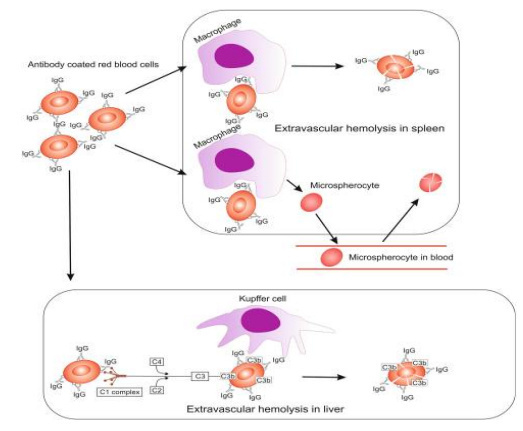
而冷抗体型AIHA(cAIHA)是由冷抗体(最适反应温度在30℃以下的自身抗体)介导的溶血性贫血,以血管内溶血为主。其溶血机制为冷凝集素与红细胞反应激活补体级联反应,导致血管内和血管外溶血:血管外溶血与单核吞噬系统的补体激活和红细胞的破坏有关,主要位置在肝脏;血管内溶血是由C5b、C6、C7、C8和C9组成膜攻击复合物(MAC)诱发溶血。
Immun Ageing. 2020;17(1):38.
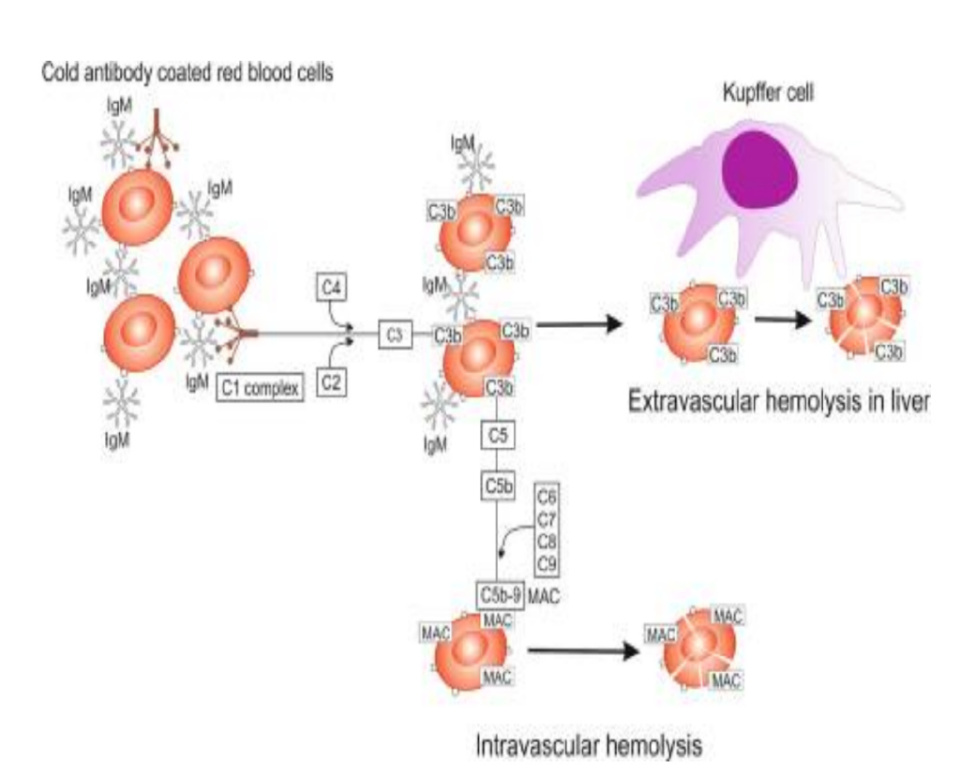
冷抗体主要为完全抗体IgM,容易聚集红细胞,可结合补体,成为冷凝集素,多见于冷凝集素病(综合征)。冷凝集素病(CAD)为原发性疾病,冷凝集素综合征(CAS)继发于其他血液系统疾病、自身免疫病或肿瘤,阵发性冷性血红蛋白尿(PCH)则常继发于病毒感染。CAD主要发病机制为克隆性B细胞增殖和补体激活,抗体类型主要为单克隆性IgM,其与红细胞表面的Ⅰ抗原结合可使红细胞发生凝集,最佳反应温度为0~4℃。IgM是一种强补体激活剂,可以与C1q结合激活经典补体途径产生C3b,当血液温度升高到37℃时,IgM从红细胞表面脱落,而C3b仍固定在红细胞上,C3b调理的红细胞被肝脏吞噬系统破坏,导致血管外溶血。
AIHA是遗传易感性与环境因素相互作用的结果,其遗传学背景复杂且多元。天津医科大学总医院针对wAIHA患者的表观遗传特征展开研究,发现患者B细胞全基因组DNA甲基化水平呈现特异性改变,C碱基和CG型碱基的总甲基化水平显著低于正常对照(P<0.05)。DNA甲基化区域(DMRs)分析显示,Rap1、细胞因子、磷脂酶D(PLD)、HIF-1、NF-κB、VEGF和Ras信号通路等相关基因的甲基化异常。此外,该中心针对AIHA的免疫机制探索发现,白细胞介素-6(IL-6)可诱导B细胞增殖和分化,提示IL-6R拮抗剂可能成为wAIHA患者的新靶向治疗方法。
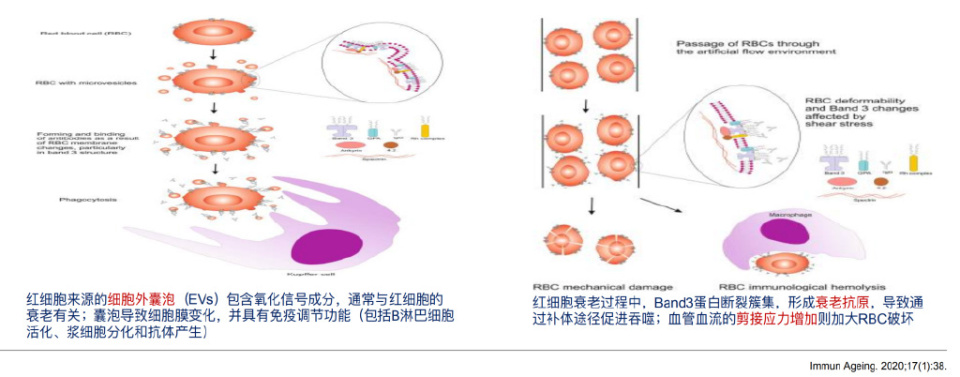
需要注意的是,AIHA的风险随着年龄的增长而增加,衰老抗原造成红细胞膜的改变导致红细胞过早死亡。衰老过程以及大量的共病增加了氧化应激,ROS水平的升高与抗红细胞自身抗体增加有关,另外ROS信号可诱导中性粒细胞细胞外陷阱(NET)的形成,影响免疫细胞功能的调节。
02、非免疫性因素
微血管病性溶血性贫血(MAHA)是⼀种因微血管病变导致红细胞在微循环中被机械性破坏而发生的溶血性贫血,其特征为外周血中出现破碎红细胞和球形细胞,常伴有血小板减少和器官损害,常见于弥散性血管内凝血(DIC)、血栓性血小板减少性紫癜(TTP)、溶血尿毒症综合征(HUS)等。此外,机械性溶血(如人工心脏瓣膜)、感染(如疟疾)及理化因素(如铅中毒)也可通过损伤红细胞膜或直接破坏红细胞引发溶血。
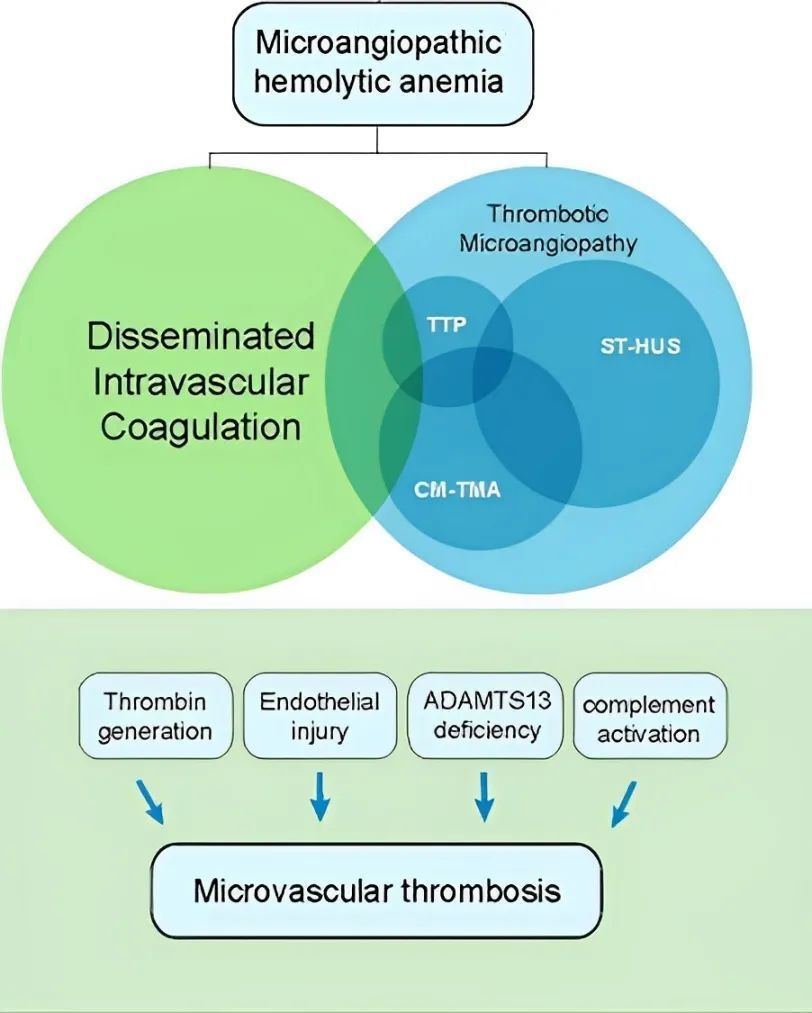
Hematology Am Soc Hematol Educ Program. 2023;2023(1):43-50.
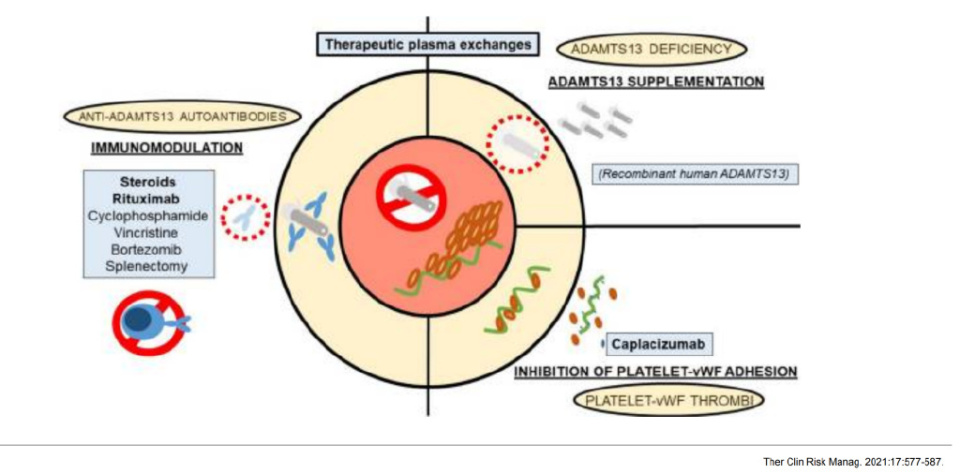
其中,TTP的发病机制主要涉及血管性血友病因子(VWF)裂解酶(ADAMTS13)活性缺乏,也与血管内皮细胞VWF异常释放、补体异常活化、血小板异常活化等相关。主要分为先天性(cTTP)和获得性(aTTP)如免疫介导的TTP(iTTP)。血浆中ADAMTS13活性缺乏导致内皮细胞异常释放的超大VWF多聚体(UL-VWF)不能及时降解,UL-VWF可自发结合血小板,导致微血管内血栓形成、微血管病性溶血,进而引起相应器官缺血、缺氧及功能障碍,引起各种临床症状。
HUS是以溶血性贫血、血小板减少及急性肾功能衰竭为特征的⼀种综合征,与TTP都属于血栓性微血管病(TMA),主要临床表现是急性肾损伤、血小板减少及急性微血管病性溶血性贫血(外周血涂片可找到红细胞碎片)。微血栓主要分布于肾脏,成人及儿童均可发病,以儿童多见,是儿童急性肾衰竭常见原因之一。
Cells. 2021;10(12):3580.
补体旁路途径调节异常是非典型溶⾎性尿毒症综合征(aHUS)发病的主要原因,分为先天性和获得性。先天性补体调控缺陷存在补体基因突变,突变基因包括C3基因、补体H因子(CFH)基因、H因子相关蛋白(CFHR)基因、补体I因子(CFI)基因、补体B因子(CFB)基因、膜辅助蛋白(MCP)基因、血栓调节蛋白(THBD)基因等。
诊断与鉴别诊断
01、实验室检查
HA的原因和类型尽管众多,但在临床中,通常将实验室检查分为两个先后进行的基本步骤:即一般性的常规项目检查和溶血试验。一般的常规性检查包括血常规检查、血片形态学检查、网织红细胞检查、骨髓检查、血液生化检查、尿常规检查、肝胆脾B超检查等基本确认溶血有无,以提示HA的一些可能或大体的病因,而对经过临床病史分析和一般的常规性检查而基本符合的HA患者,从确定溶血以及大体评判溶血的程度、确立发生溶血的主要场所、寻找溶血的病因或原因三个方面有选择地进行溶血试验以进一步确定。
介导AIHA的不完全抗体(以IgG为主)或补体分子(以C3为主),虽然能与红细胞结合(致敏红细胞),但不能直接引起红细胞凝集。Coombs试验根据检测的是结合于红细胞上的还是游离于血清中的自身抗体而分为直接Coombs试验和间接Coombs试验。直接Coombs试验(DAT)可同时测定吸附在红细胞膜上的不完全抗体和补体,是AIHA诊断的重要指标。受检致敏红细胞经洗涤后加入抗人球蛋白抗体,抗人球蛋白抗体与不完全抗体Fc段相结合,起到“搭桥”作用而使致敏红细胞发生凝集,即直接Coombs试验阳性。根据加入的抗人球蛋白不同,可鉴别使红细胞致敏的是IgG抗体还是补体C3或IgG+C3复合型。间接Coombs试验(IAT)用于检测游离在血清中的不完全抗体。
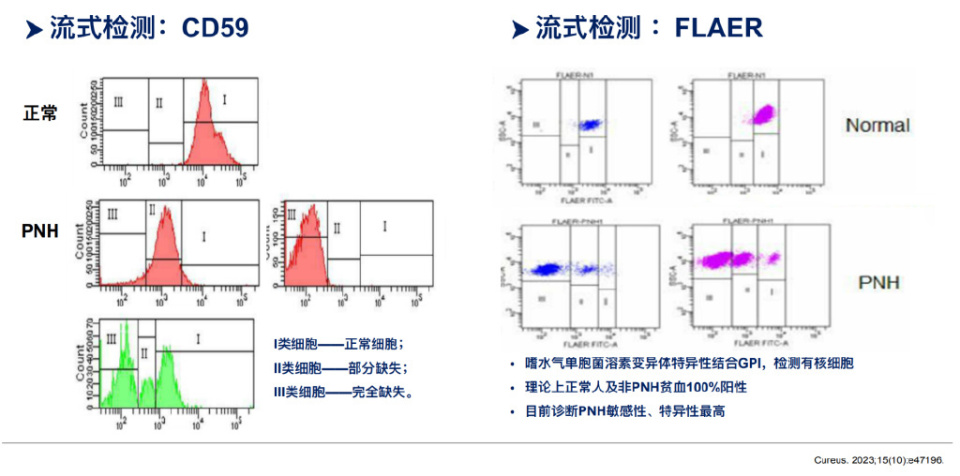
在PNH疾病进程中,使用流式细胞术检测红细胞和白细胞表面GPI锚定蛋白(如CD55和CD59)的表达情况;PNH患者的检测结果会显示部分或全部红细胞和白细胞缺乏CD55和CD59表达,从而产生弱或无荧光信号。一般来说,PNH克隆比例在患者中为0.01%到100%之间,健康个体的PNH克隆比例应为0%。使用FLARE检测更灵敏地检测低水平的GPI锚定蛋白表达;FLARE检测的正常值应为0%,即所有细胞均显示GPI锚定蛋白的强表达。PNH患者的FLARE检测结果会显示不同程度的GPI锚定蛋白缺失,根据荧光信号的强度,可以确定PNH克隆的比例。
02、筛查PNH克隆的指征
1.以血红蛋白尿、尿含铁血黄素阳性和(或)血清游离血红蛋白增高为主要表现的血管内溶血。
2.无法解释的溶血伴有腹痛或食管痉挛、血栓栓塞、血小板减少和(或)白细胞减少。
3.Coombs试验(-)、无明显肝脾肿大、极少见红细胞碎片、非感染性溶血性贫血。
4.骨髓衰竭症:
①怀疑或确诊的再生障碍性贫血或低增生性贫血;
②难治性血细胞减少伴一系或多系发育异常;
③不明原因的血细胞减少症。
5.不同寻常的血栓形成:
①非常见部位血栓形成:肝静脉(Budd-Chiari综合征)、其他腹腔内静脉(门静脉、脾静脉等)、海绵窦、皮肤静脉;
②伴有溶血征象的血栓形成;
③伴有全血细胞减少的血栓形成。
03、HA鉴别诊断
溶血性贫血需与多种相似表现的疾病鉴别,根据症状(如贫血症状)、体征(如脾大、黄疸)、采集的病史(如家族史、服药史和其他病史),以及实验室检查有易于破坏的红细胞(如球形、靶形、椭圆形红细胞)或已经破坏的红细胞(如盔形、三角形和其他破坏的红细胞)和(或)红细胞破坏增加(如网织红细胞增加、间接胆红素增加)、红系代偿性造血(骨髓有核红细胞增加)和(或)红细胞缺陷(如G-6-PD缺乏、丙酮酸激酶缺乏)几个方面的依据并有贫血者,即可以确诊HA。
诊断时,先通过LDH、结合珠蛋白、间接胆红素、RET、尿胆原等明确是否存在溶血,再根据血涂片红细胞形态、DAT试验、G-6-PD活性检测、基因检测等结果,结合病史(用药史、感染史等)进行综合判断,区分不同类型的溶血性贫血。
总结
溶血性贫血包括遗传性和获得性,诊断需要综合病史、家族史、体格检查和实验室检查等多方面信息。鉴别诊断对于明确病因、采取针对性治疗至关重要,需依据溶血部位和实验室检查结果仔细鉴别。早期诊断和及时治疗对于避免严重并发症、提高治愈率、改善患者预后意义重大。对PNH、AIHA、β地中海贫血等特殊亚型发病机制的深入研究,有助于开发新的治疗靶点,推动溶血性贫血治疗的发展。

付蓉 教授
天津医科大学总医院
医学博士、主任医师、二级教授、博士生导师
天津医科大学总医院 副院长、血液病中心 主任
天津市骨髓衰竭及癌性造血克隆防治重点实验室 主任
天津市血液病研究所 所长
中华医学会血液学分会 常委
中国医师协会血液科医师分会 常委
中华医学会血液学分会红细胞学组 组长
北京癌症防治学会红细胞疾病专委会 主委
中国抗癌协会血液肿瘤专业委员会中国MDS/MPN工作组 副组长
中国临床肿瘤学会(CSCO)骨髓瘤专业委员会 常委
中国女医师协会临床肿瘤学专业委员会 常委
中国医学协会血液学机构分会 副主委
天津市医学会血液学分会 主委
天津市医师协会血液科医师分会 副会长
天津市医疗健康学会血液病学专业委员会 主任委员
Journal of clinical Laboratory Analysis (SCI)主编
中华血液学杂志 副总编
首届天津名医、天津市教学名师、天津科普大使、津门医学英才
主笔《AA诊断与治疗中国指南》、《PNH诊断与治疗中国指南》、《PRCA诊断与治疗中国专家共识》等